
 El Sistema del Complemento (CS) es un conjunto de proteínas séricas que actúan de manera altamente controlada en diversos tipos de respuestas inmunoinflamatorias con anticuerpos. El complemento fue reconocido a finales del siglo XIX como una de las 2 proteínas saludables solubles en el producto sanguíneo humano a cargo de la mortalidad de las bacterias, siendo el otro compuesto el anticuerpo. La proteína potenciadora original se denominó Alexina, sin embargo, su nombre finalmente se cambió para mostrar exactamente cómo la proteína „potenciaba” la acción del anticuerpo para llevar a cabo la lisis microbiana. Enhance está formado por proteínas sanas solubles en plasma expresadas en la capa de membrana celular y se desencadena por una serie de mecanismos con tres vías: la vía atemporal, la vía diferente y la vía de lectina. Estas 3 vías de activación mejoradas difieren en la forma en que se lanzan, pero comparten acciones finales, ejecutando las mismas características de efecto. La opción, así como las vías de lectina, son mecanismos de efecto de la inmunidad natural, mientras que la vía clásica se encuentra entre los principales sistemas de inmunidad humoral flexible. SC se une a la fagocitosis, opsonización, quimiotaxis de leucocitos, liberación de histamina de mastocitos y basófilos y también variedades energéticas de oxígeno de leucocitos, vasoconstricción, endurecimiento de la masa muscular lisa, permeabilidad mejorada de los vasos, recolección de plaquetas y citólisis. El sistema del complemento es uno de los sistemas principales mediante los cuales el reconocimiento del virus se convierte en una reacción inmunitaria eficaz contra una infección preliminar. Enhance es un sistema de proteínas plasmáticas que puede activarse directamente por virus o indirectamente con anticuerpos unidos a microorganismos, lo que lleva a una cascada de respuestas que tienen lugar externamente al virus y crean elementos activos con diversas funciones de efecto.
El Sistema del Complemento (CS) es un conjunto de proteínas séricas que actúan de manera altamente controlada en diversos tipos de respuestas inmunoinflamatorias con anticuerpos. El complemento fue reconocido a finales del siglo XIX como una de las 2 proteínas saludables solubles en el producto sanguíneo humano a cargo de la mortalidad de las bacterias, siendo el otro compuesto el anticuerpo. La proteína potenciadora original se denominó Alexina, sin embargo, su nombre finalmente se cambió para mostrar exactamente cómo la proteína „potenciaba” la acción del anticuerpo para llevar a cabo la lisis microbiana. Enhance está formado por proteínas sanas solubles en plasma expresadas en la capa de membrana celular y se desencadena por una serie de mecanismos con tres vías: la vía atemporal, la vía diferente y la vía de lectina. Estas 3 vías de activación mejoradas difieren en la forma en que se lanzan, pero comparten acciones finales, ejecutando las mismas características de efecto. La opción, así como las vías de lectina, son mecanismos de efecto de la inmunidad natural, mientras que la vía clásica se encuentra entre los principales sistemas de inmunidad humoral flexible. SC se une a la fagocitosis, opsonización, quimiotaxis de leucocitos, liberación de histamina de mastocitos y basófilos y también variedades energéticas de oxígeno de leucocitos, vasoconstricción, endurecimiento de la masa muscular lisa, permeabilidad mejorada de los vasos, recolección de plaquetas y citólisis. El sistema del complemento es uno de los sistemas principales mediante los cuales el reconocimiento del virus se convierte en una reacción inmunitaria eficaz contra una infección preliminar. Enhance es un sistema de proteínas plasmáticas que puede activarse directamente por virus o indirectamente con anticuerpos unidos a microorganismos, lo que lleva a una cascada de respuestas que tienen lugar externamente al virus y crean elementos activos con diversas funciones de efecto.
Cómo funciona el sistema complementario?
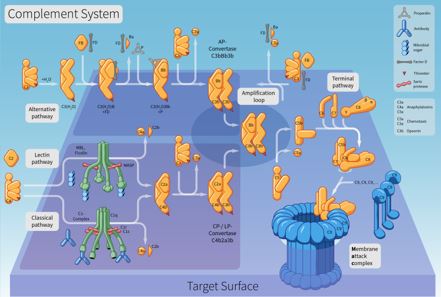
 El sistema del Complemento consiste en aproximadamente 35 proteínas y también glicoproteínas distribuidas en plasma, otros líquidos biológicos y también áreas de superficie celular. Estas proteínas saludables actúan en una reacción en cascada consecutiva y también representan uno de los principales conciliadores de la defensa e inflamación innatas del huésped. La activación del sistema del complemento puede ser beneficiosa para el huésped, tal como la resistencia a los microbios atacantes, y también negativa en una variedad de afecciones mediadas inmunopatológicamente. la potenciación puede activarse mediante una cualquiera de las tres rutas, la ruta atemporal dependiente de anticuerpos, la ruta diferente o la ruta de lectina (MBL) explicada recientemente. Las consecuencias orgánicas de la activación del complemento son principalmente la protección frente a infecciones piógenas, la interacción entre la inmunidad innata y adaptativa, así como la eliminación de complejos inmunes y elementos de lesión celular.
El sistema del Complemento consiste en aproximadamente 35 proteínas y también glicoproteínas distribuidas en plasma, otros líquidos biológicos y también áreas de superficie celular. Estas proteínas saludables actúan en una reacción en cascada consecutiva y también representan uno de los principales conciliadores de la defensa e inflamación innatas del huésped. La activación del sistema del complemento puede ser beneficiosa para el huésped, tal como la resistencia a los microbios atacantes, y también negativa en una variedad de afecciones mediadas inmunopatológicamente. la potenciación puede activarse mediante una cualquiera de las tres rutas, la ruta atemporal dependiente de anticuerpos, la ruta diferente o la ruta de lectina (MBL) explicada recientemente. Las consecuencias orgánicas de la activación del complemento son principalmente la protección frente a infecciones piógenas, la interacción entre la inmunidad innata y adaptativa, así como la eliminación de complejos inmunes y elementos de lesión celular.
Enhance reconoce, opsoniza o lisa partículas, incluidos microorganismos, levaduras, así como otras bacterias diversas, desechos móviles, así como cambios en el huésped celular. También hay evidencia de que la mejora se suma significativamente a la ley de la retroalimentación inmune. El objetivo principal de esta revisión es ofrecer una visión existente de la activación y también de los hogares orgánicos de este sistema, enfatizando su relevancia en la reacción inmune inherente y también en la homeostasis del huésped. El sistema enhance es difusamente activo en el cuerpo y la escasez o desregulación conduce a la escasez del sistema inmunológico del cuerpo, enfermedades autoinmunes o problemas de hemorragia. El sistema enhance juega un papel crucial en la inflamación, así como en la defensa contra algunas infecciones bacterianas. El complemento también podría desencadenarse durante las respuestas frente a las transfusiones de sangre inapropiadas y también durante las reacciones inmunitarias peligrosas que acompañan a la enfermedad autoinmune. Debido a que no se requieren anticuerpos para activar la ruta alternativa, sin embargo, se necesitan para activar la cascada clásica, la ruta diferente actúa como una protección inicial contra la infección, así como pertenece a la respuesta inmune innata inespecífica, que tiene lugar antes de que se pueda instalar una reacción inmune adquirida particular. La ruta diferente parece ser una de las más primitivas de los 3 sistemas.
Relevancia del Sistema del Complemento
 El sistema potenciador ayuda o „complementa” la capacidad de los anticuerpos y las células fagocíticas para eliminar microorganismos de un microorganismo. Pertenece al sistema inmune innato del cuerpo. El sistema de mejora se compone de una colección de pequeñas proteínas saludables ubicadas en la sangre, producidas por el hígado. Generalmente se distribuyen como precursores inactivos. Cuando son promovidas por un desencadenante, las proteasas dividen estas pequeñas proteínas para liberar citocinas activas. El sistema de mejora es un componente importante de la respuesta inmune inherente y también funciona como un puente entre la resistencia inherente y adquirida.
El sistema potenciador ayuda o „complementa” la capacidad de los anticuerpos y las células fagocíticas para eliminar microorganismos de un microorganismo. Pertenece al sistema inmune innato del cuerpo. El sistema de mejora se compone de una colección de pequeñas proteínas saludables ubicadas en la sangre, producidas por el hígado. Generalmente se distribuyen como precursores inactivos. Cuando son promovidas por un desencadenante, las proteasas dividen estas pequeñas proteínas para liberar citocinas activas. El sistema de mejora es un componente importante de la respuesta inmune inherente y también funciona como un puente entre la resistencia inherente y adquirida.
Incluye una serie de proteínas que se sintetizan básicamente (aunque no únicamente) en el hígado que existen en el plasma y también en el área de la superficie celular como precursores no activos (zimógenos). Esto comienza una serie (una cascada) de senos adicionales que liberan aún más citocinas. Esto magnifica la reacción. Por lo tanto, si la estimulación inicial fue un microorganismo atacante, las citocinas alteran la membrana celular bicapa de fosfolípidos del objetivo, lo que lo mata. Los suplementos son proteínas solubles y glicoproteínas, más de 20 tipos de suplementos están presentes en el suero, descubiertos circulando generalmente en el cuerpo en tipos inactivos. Los complementos se activan solo a lo largo de las reacciones inflamatorias. Durante la inflamación, más enhance llega a la ubicación intersticial de las células contaminadas a través de vasos sanguíneos dilatados, que luego se activan por escisión proteolítica. Las proteínas saludables del complemento en el flujo no se activan hasta que se activan por un encuentro con una célula microbiana, una infección, una célula inmune complicada, dañada u otro material que normalmente no está presente en el cuerpo. La activación del complemento es una ocasión de inmersión, como el otoño de una fila de fichas de dominó. Debe seguir un orden de detalles para asegurarse de que se logre el resultado. El sistema de mejora tiene la posibilidad de ser muy dañino para las células huésped; por esa razón, se requieren sistemas reguladores para limitar la ruta del complemento. Numerosas proteínas plasmáticas, así como de la capa de membrana celular, controlan la activación al inhibir diferentes acciones de la Cascada.